GeneticMapTool
制作:张敖 https://datahold.cn
鸣谢
张学才,CIMMYT,指导
最新更新
【2020-10-06】
基本功能实现。
功能
- 利用遗传距离信息绘制遗传图谱的“蜈蚣图”和密度图,PDF格式。
- 本程序使用LinkageMapView包,程序运行后可以自行根据LinkageMapView包的函数个性化制作。
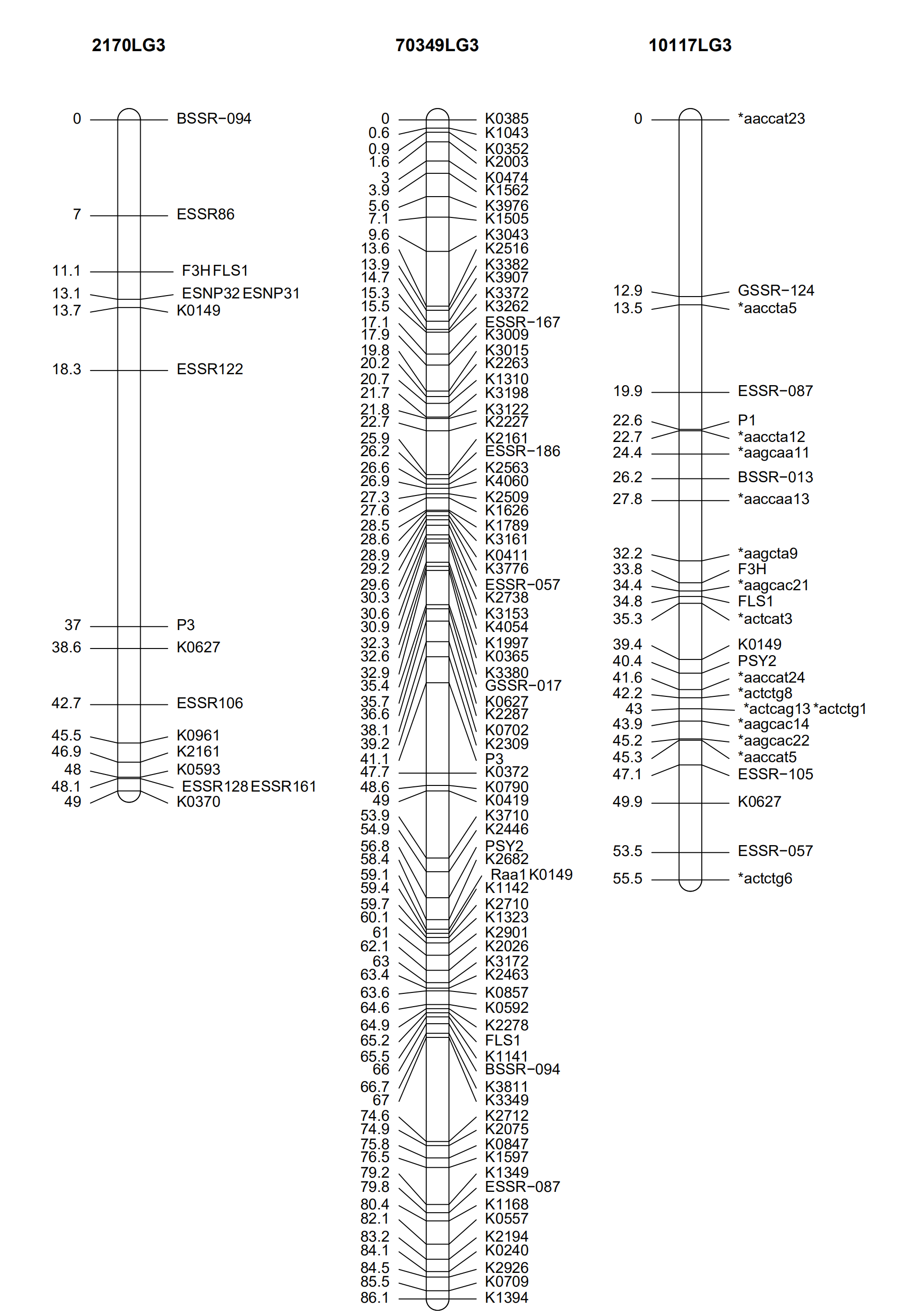
生成图展示
蜈蚣图展示
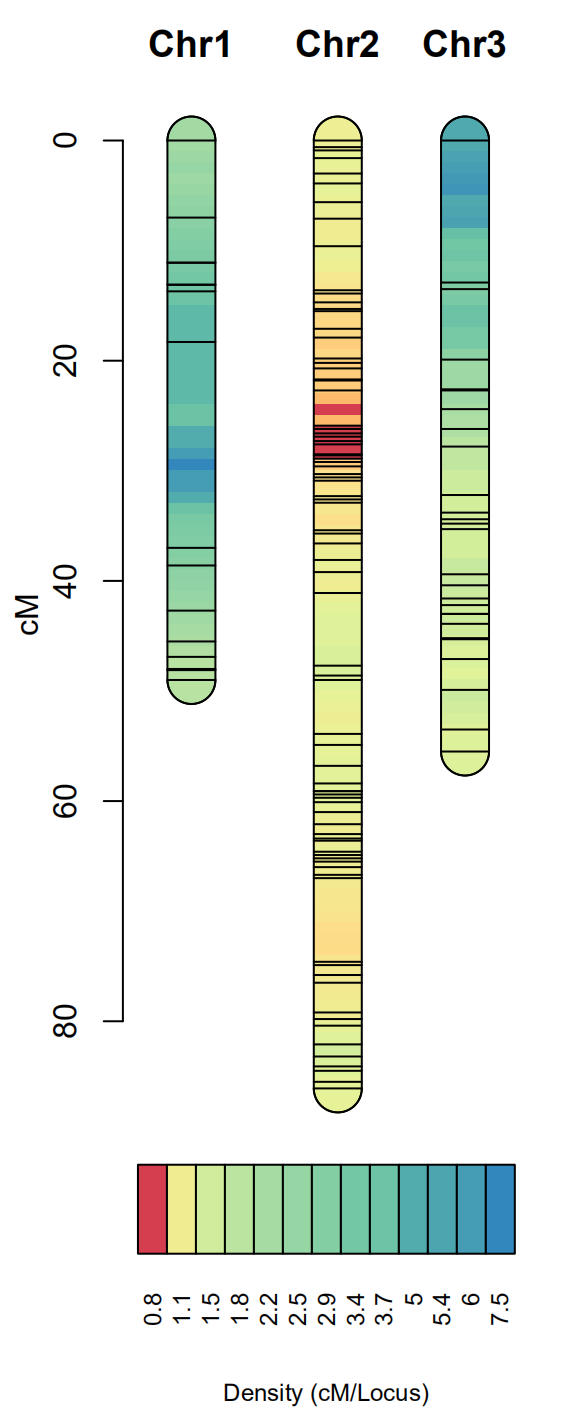
密度图展示
输入文件展示
输入文件按照下列格式准备,第一列为染色体列,第二列为遗传距离(单位cM),可以使用各种软件计算遗传距离后填入表中,第三列为标记列。【示例下载】
| Chr | position | locus |
|---|---|---|
| Chr1 | 0 | BSSR-094 |
| Chr1 | 7.039 | ESSR86 |
| Chr1 | 11.123 | F3H |
| Chr1 | 11.123 | FLS1 |
| Chr1 | 13.079 | ESNP32 |
| Chr1 | 13.079 | ESNP31 |
| Chr1 | 13.734 | K0149 |
| Chr1 | 18.304 | ESSR122 |
| Chr1 | 36.975 | P3 |
| Chr1 | 38.576 | K0627 |
| Chr1 | 42.699 | ESSR106 |
| Chr1 | 45.453 | K0961 |
| Chr1 | 46.876 | K2161 |
| Chr1 | 48.033 | K0593 |
| Chr1 | 48.075 | ESSR128 |
| Chr1 | 48.075 | ESSR161 |
| Chr1 | 48.965 | K0370 |
| Chr2 | 0 | K0385 |
使用方法
准备好输入文件,然后在R Studio中运行下列代码。
海外:
x
rm(list=ls()) # Clear the Environment Variables 清空环境变量 source("https://aozhangchina.github.io/R/GeneticMapTool/GeneticMapTool.R") # 加载程序文件,需要联网国内:
x
rm(list=ls()) # Clear the Environment Variables 清空环境变量 source("https://dataholdcn.cn/R/GeneticMapTool/GeneticMapTool.R") # 加载程序文件,需要联网需要选择准备好的文件。
程序自动运行直至全部完毕。